






Berkeley Researchers Crack Open ‘AI-at-Scale’ Method for Chemical Science
December 11, 2024
By David Krause
Contact: cscomms@lbl.gov
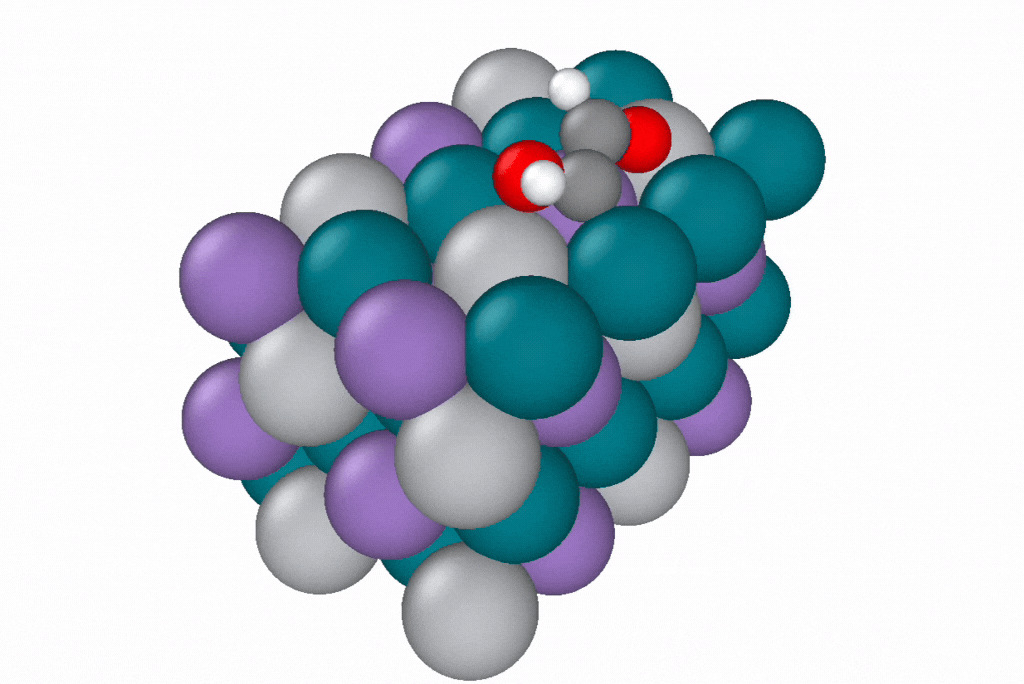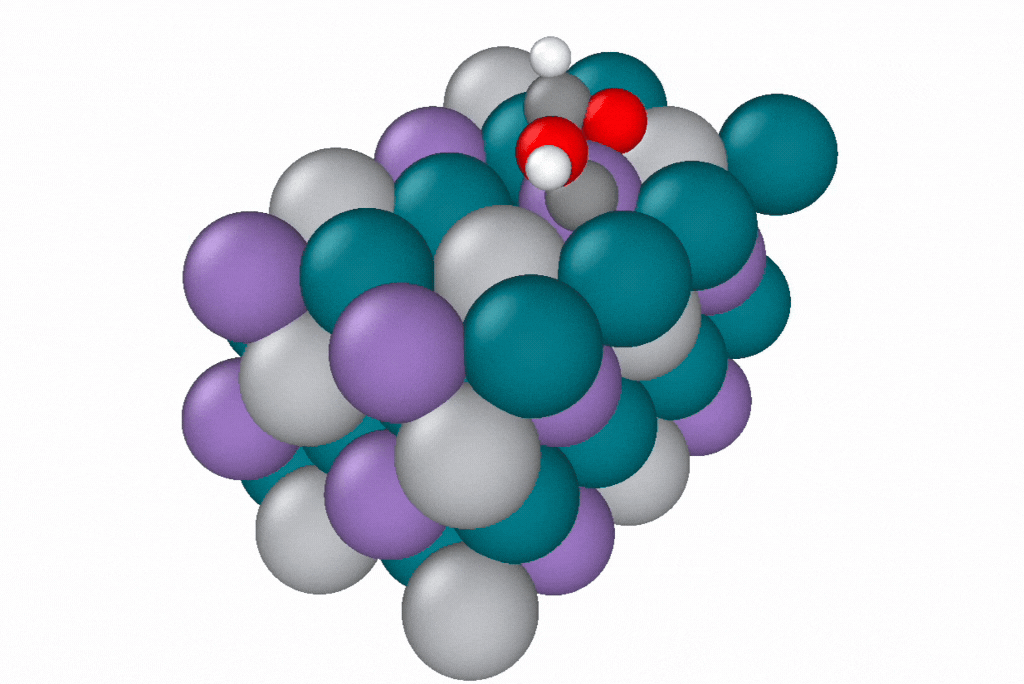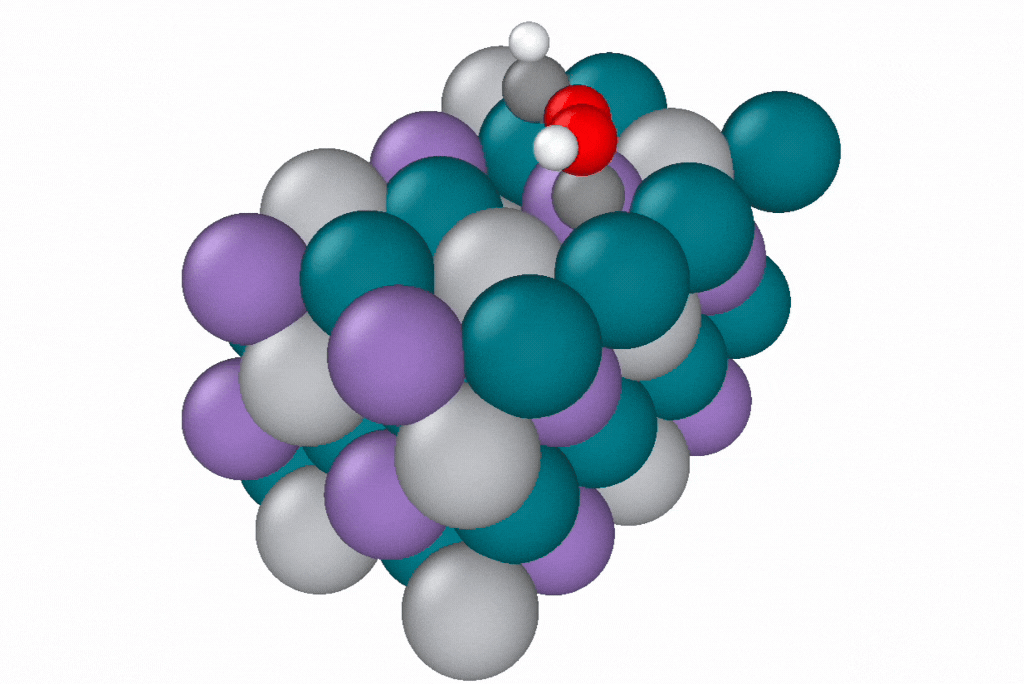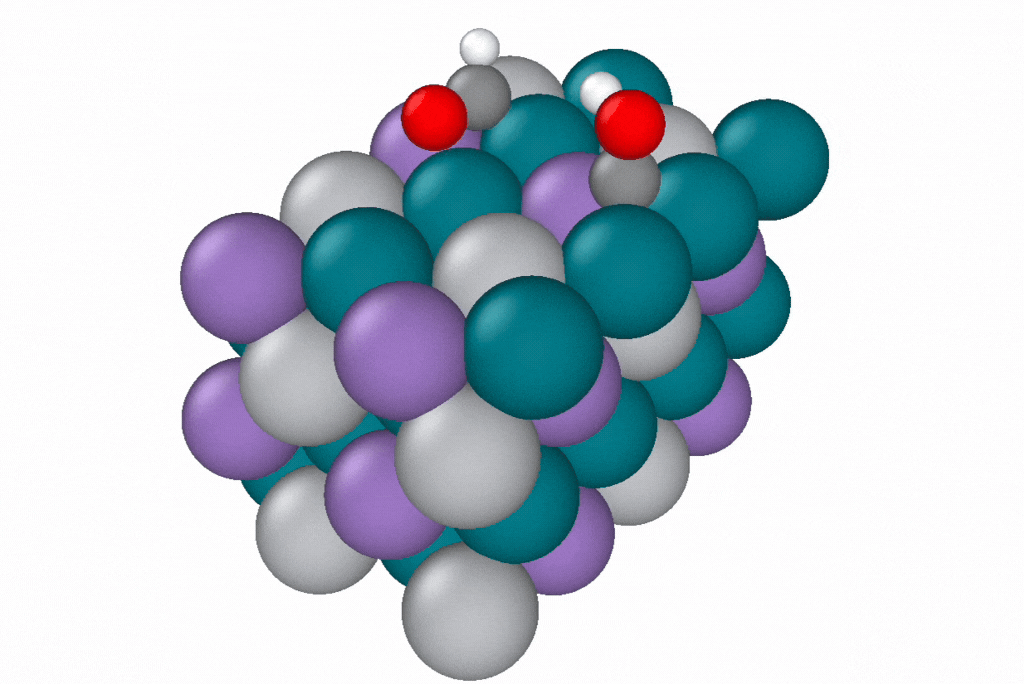
A reaction path generated by a novel path optimization method, under development by Samuel Blau and co-workers, using EScAIP trained on Open Catalyst Project data. EScAIP's speed and low memory cost are critical for optimizing reaction paths of structures containing many atoms. Image courtesy of Samuel Blau and Eric Yuan, Berkeley Lab.
Quantum calculations of molecular systems often require extraordinary amounts of computing power; these calculations are typically performed on the world’s largest supercomputers to better understand real-world products such as batteries and semiconductors.
Now, UC Berkeley and Lawrence Berkeley National Laboratory (Berkeley Lab) researchers have developed a new machine learning method that significantly speeds up atomistic simulations by improving model scalability. This approach reduces the computing memory required for simulations by more than fivefold compared to existing models and delivers results over ten times faster. Their research has been accepted at Neural Information Processing Systems (NeurIPS) 2024, one of the premier conferences and publication venues in artificial intelligence and machine learning. They will present their work at the conference on December 13.
“We wanted to build a different kind of machine learning architecture using methods typically applied to large language models,” said Eric Qu, a UC Berkeley graduate student and co-author of the research paper. “With our approach, researchers can more efficiently map how atoms move around and interact with each other.”
Understanding what happens to nature’s smallest building blocks can open a deeper understanding of materials science, chemistry, and drug development, among other basic science subjects.
“This model can help scientists determine chemical reaction mechanisms much more efficiently,” said Samuel Blau, a Berkeley Lab computational chemist. “If you can understand the complex chemistry in real-world systems, you can figure out how to control them in new ways.”
Hungry Scales
Over the past decade, scientists and engineers have built large language models like ChatGPT using massive datasets and a strategy called scaling. Scaling involves making these models bigger and smarter by systematically increasing the number of parameters in the neural networks. How you increase these parameters matters: different parameters contribute to model performance in distinct ways, and optimizing this process can lead to significant improvements. Researchers can also design new operations or components within the neural network architecture—such as novel attention mechanisms—that are more expressive, enabling further increases in parameters while maintaining or improving efficiency. But it’s not just about size; scaling also means finding ways to make these models more efficient, using smarter algorithms to save time and computing power during both training and use. Instead of focusing solely on raw processing power, researchers often measure efficiency by how long it actually takes to train or run these models, prioritizing real-world performance.
However, the principles of scaling have not been extensively applied to another type of machine learning model particularly useful for scientists: Neural Network Interatomic Potentials (NNIPs). NNIPs serve as efficient alternatives to computationally expensive quantum mechanical simulations that allow researchers to predict molecular and material properties much faster.
“NNIPs are rapidly becoming the most powerful approach for molecular or materials simulation,” said Aditi Krishnapriyan, paper co-author, UC Berkeley assistant professor, and faculty scientist in Berkeley Lab’s Applied Mathematics and Computational Research Division. “Previously, designing clever algorithms at scale was primarily being developed in other fields of machine learning, such as large language models, and less so for studying materials, chemistry, or physics.”
The Berkeley team thus developed an NNIP architecture that could be scaled effectively. The architecture, known as Efficiently Scaled Attention Interatomic Potential (EScAIP), represents a significant step forward for scaling machine learning models for scientific applications, Krishnapriyan said.
Raining Data
While large language models such as ChatGPT are trained on text, for which trillions of examples exist on the internet, NNIPs rely on data generated by a technique common in computational research called density functional theory (DFT). DFT is a physics-based numerical approach that uses quantum mechanics to predict how atoms interact in molecules and materials. Although DFT simulations are very powerful, they are also computationally expensive, and generating a large amount of DFT training data can be very time-consuming. Machine learning has the potential to accelerate these simulations by acting as a surrogate model for DFT. Only recently have DFT datasets with 100 million data points been released, with previous datasets maxing out at around 1-2 million, setting the stage for scaling to be crucial for NNIPs.
However, current NNIP models that incorporate physical constraints often require substantial computer hardware, memory, and processing time, and it can also add complications in optimizing the neural network parameters easily. In contrast, EScAIP does not include a lot of built-in physical constraints; instead, it focuses on making the machine learning model as expressive as possible, including by designing a new attention mechanism customized for the atomistic setting. This approach enables EScAIP to capture complex patterns in the data and learn key physical insights directly from the data itself, bypassing the need for explicit constraints. For example, after training, EScAIP can, on new, unseen atomic systems, accurately map any atomic orientation to predicted forces, capturing a symmetry known as rotational equivariance.
“The new EScAIP model can train on 100 million data points in a matter of days, whereas a physically-constrained NNIP would require weeks or months,” Blau said. As a result, the number of research groups that can feasibly train these models expands dramatically.
“We really believe in helping people pursue their science goals using tools previously less accessible,” Qu added. “EScAIP gives scientists with different amounts of resources that chance.”
EScAIP is a significant improvement over previously state-of-the-art NNIPs, with trained models achieving the top performance on common NNIP benchmark datasets spanning diverse chemical systems, including catalysts (such as the Open Catalyst Project), materials (such as the Materials Project), and molecules (such as SPICE). On datasets like Open Catalyst, it is also the first model to top the leaderboard that was developed and trained purely by academic and national lab researchers, rather than by teams at major technology companies. However, Qu and Krishnapriyan believe that the model should be viewed as a first step in a new direction.
“We are saying to the science community, ‘Hey, look over here, let’s explore this idea more,’” Krishnapriyan said. “EScAIP is an initial proof-of-concept for how to think about scaling machine learning models in the context of atomistic systems, and now represents a “lower bound” for what’s possible. We think it’s the direction that we should be thinking about going in the field as we enter a future with more data and computational resources.”
According to Krishnapriyan, EScAIP traces its origins to a Berkeley Lab Laboratory Directed Research and Development (LDRD) project, Development of New Physics-Informed Machine Learning Methods, which helped shape its foundational ideas. She emphasizes that leveraging the extensive GPU resources at the Department of Energy’s (DOE) National Energy Research Scientific Computing Center (NERSC) was crucial for developing and training models on large-scale datasets. By utilizing multiple GPUs simultaneously, the team achieved top performance on the Open Catalyst dataset—a remarkable accomplishment, particularly as the only non-tech company team to do so with significantly fewer resources. NERSC, a DOE user facility, is located at Berkeley Lab.
About Berkeley Lab
Founded in 1931 on the belief that the biggest scientific challenges are best addressed by teams, Lawrence Berkeley National Laboratory and its scientists have been recognized with 16 Nobel Prizes. Today, Berkeley Lab researchers develop sustainable energy and environmental solutions, create useful new materials, advance the frontiers of computing, and probe the mysteries of life, matter, and the universe. Scientists from around the world rely on the Lab’s facilities for their own discovery science. Berkeley Lab is a multiprogram national laboratory, managed by the University of California for the U.S. Department of Energy’s Office of Science.
DOE’s Office of Science is the single largest supporter of basic research in the physical sciences in the United States, and is working to address some of the most pressing challenges of our time. For more information, please visit energy.gov/science.